

Abstract
Introduction: Innate and acquired resistance to anti-cancer therapies poses a major hurdle in effectively treating many cancers, especially an incurable cancer like multiple myeloma (MM). Rational combination therapies have shown improved efficacy and reduced toxicity in MM. Patient variability in response to single agents leads to variability in combination effects, which require quantification on a patient-to-patient basis. Conventional combination effect quantification methods rely on dose - response curves obtained from experiments involving cell lines. Such studies don't account for intratumoral and intertumoral heterogeneity that play an important factor in driving a patient's clinical response.
Materials and Methods: We propose a framework that captures tumor-specific two-way combination effect in an ex vivo reconstruction of the tumor microenvironment using patient-derived primary multiple myeloma cells. The framework translates the data obtained from an ex vivo drug sensitivity assay to patient-specific combination therapy response predictions using mathematical modeling. MM cells (CD138+) extracted from fresh bone marrow aspirates are seeded in an ex vivo co-culture model with human stroma in multi-well plates, and tested with various drugs/combinations at several concentrations. Each well is imaged for at least 96 hours, once every 30 minutes to estimate percent viable cells. Such a platform facilitates measuring response with respect to dose and time, making this an ideal paradigm to capture pharmacodynamical interactions between drugs. An empirical mathematical model is used to measure the combination effects between two drugs, and when combined with their pharmacokinetic data obtained from Phase-I clinical trials the model predicts patient-specific response over a 90 day treatment period within five days post biopsy.
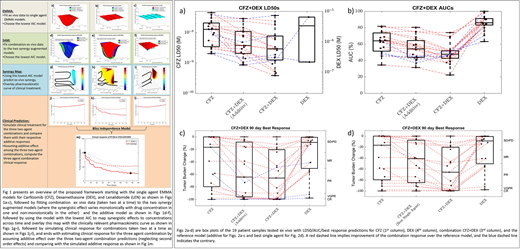
Results: A total of 58 multiple myeloma patient samples were tested ex vivo with 19 two-drug combinations. The resulting ex vivo response data is fit to single agent (EMMA - Ex vivo Mathematical Myeloma Advisor) and combination (SAM - Synergy Augmented Model) mathematical models to estimate patient-drug/combination-specific LD50s and area under the curves (AUCs) from the dose-time-response curves (shown in Figs. 1a-f). The 96 hour single agent, additive (in the sense of Bliss), and combination LD50s for 19 patients tested with the combination Carfilzomib and Dexamethasone (CFZ+DEX) are presented as a box plot in Fig. 2a . A red dashed line signifies a patient who would see a benefit over additive LD50 (synergism), while a blue dashed line implies the opposite (antagonism). Similarly, Fig. 2b presents the AUCs as a box plot, where the "area" in AUC is in fact the volume under the dose-time-response curve. Inclusion of the time axis accounts for exposure-response effect in addition to the dose-response effect captured in LD50. The effect of accounting for exposure via AUC suggests greater synergy than LD50 as seen in Figs. 2a-b. In spite of being insightful, a decrease in LD50 and/or AUC doesn't always translate to a synergistic effect in patients. In order to predict the response observed in patients, the ex vivo models are integrated with pharmacokinetic data from Phase-I clinical trials to simulate patients' response over a 90 day treatment period (shown in Figs. 1j-l). The best response over a 90 day period for the single agents, additive, and the combination are presented in Fig. 2c as a box plot and the right y-axis classifies the response. However, additive effect is a theoretically computed quantity that may have pharmacological relevance but isn't significant clinically. A more clinically relevant reference model would be to compare the combination response with the better of the two single agents. Figure 2d presents the box plot comparing the predicted best single agent and combination responses. The model predictions indicate all of the 19 patients would benefit from the combination, although the extent of benefit varies from patient-to-patient.
Conclusion: The proposed framework captures patient-specific combination effects using a pharmacodynamic model that can be used to screen for the most efficacious combination for a patient and across a cohort.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal